Kawasaki disease
| Kawasaki disease | |
|---|---|
| Classification and external resources | |
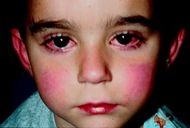 A child showing characteristic lip changes, and red eyes seen in Kawasaki disease |
|
| ICD-10 | M30.3 |
| ICD-9 | 446.1 |
| OMIM | 611775 |
| DiseasesDB | 7121 |
| MedlinePlus | 000989 |
| eMedicine | ped/1236 |
| MeSH | D009080 |
Kawasaki disease (KD), also known as Kawaski syndrome, lymph node syndrome and Mucocutaneous lymph node syndrome,[1] is an autoimmune disease that manifests as a systemic necrotizing medium-sized vessel vasculitis and is largely seen in children under 5 years of age. It affects many organ systems, mainly those including the blood vessels, skin, mucous membranes and lymph nodes; however, its most serious effect is on the heart where it can cause severe coronary artery aneurysms in untreated children. Without treatment, mortality may approach 1%, usually within 6 weeks of onset. With treatment, the mortality rate is less than 0.01% in the U.S.[2] There is often a pre-existing viral infection that may play a role in its pathogenesis.[3] The conjunctival and oral mucosa, along with the epidermis (skin), become erythematous (red and inflamed). Edema is often seen in the hands and feet and the cervical lymph nodes are often enlarged. Also, a remittent fever, often 40℃ (104°F) or higher, is characteristic of the acute phase of the disease.[4]In untreated children, the febrile period lasts on average approximately ten days, but may range from 5 to 25 days.[4] The disorder was first described in 1967 by Dr. Tomisaku Kawasaki in Japan.[5]
Contents |
Classification
Systemic vasculitis is an inflammatory condition affecting both veins and arteries throughout the body, and is usually caused by a proliferation of cells associated with an immune response to a pathogen, or autoimmunity.[6] Systemic vasculitides may be classified according to the type of cells involved in the proliferation, as well as the specific type of tissue damage occurring within the vein or arterial walls.[6] Under this classification scheme for systemic vasculitis, Kawasaki disease is considered to be a necrotizing vasculitis (also called necrotizing angeititis), which may be identified histologically by the occurrence of necrosis (tissue death), fibrosis, and proliferation of cells associated with inflammation in the inner layer of the vascular wall.[6][7] Other diseases featuring necrotizing vasculitis include Polyarteritis nodosa, Wegener's granulomatosis, Henoch-Schönlein purpura and Churg-Strauss syndrome.[6] Kawasaki disease may be further classified as a medium-sized-vessel vasculitis, affecting medium and small sized blood vessels,[8][9][10] such as the smaller cutaneous vasculature (veins and arteries in the skin) that range from 50 to 100µm in diameter.[11][12] KD is also considered to be a primary childhood vasculitis, a disorder associated with vasculitis that mainly affects children under the age of 18.[13][14] A recent, consensus-based evaluation of vasculitides occurring primarily in children resulted in a classification scheme for these disorders, to both distinguish them and suggest a more concrete set of diagnostic criteria for each.[14] Within this classification of childhood vasculitides, Kawasaki disease is, again, a predominantly medium-sized vessel vasculitis.[14]
It is also an autoimmune form of vasculitis,[4] and is not associated with ANCA antibodies, unlike other vasculitic disorders associated with them, such as wegener's granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome.[6][15] This categorization is considered essential for appropriate treatment.[16]
Signs and symptoms
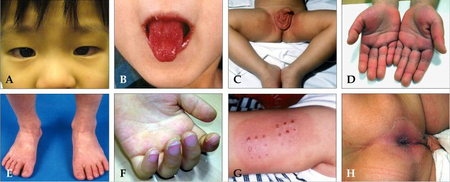
Kawasaki disease often begins with a high and persistent fever that is not very responsive to normal treatment with paracetamol (acetaminophen) or ibuprofen.[17][18] The fever may persist steadily for up to two weeks and is normally accompanied by irritability.[18][17] Affected children develop red eyes because of non-suppurative conjunctivitis, iritis[19] and bilateral anterior uveitis.[20] Inflammation of the mucous membranes in the mouth,[4] along with erythema (redness), edema (swelling) with fissures (cracks in the lip surface), desquamation (peeling) and exsudation of the lips are also evident. The oropharynx mucosa has enanthema and the tongue maintains an unusual red appearance termed "strawberry tongue" (marked erythema with prominent gustative papillae).[12] Keratic precipitates (detectable by a slit lamp but usually too small to be seen by the unaided eye), and swollen lymph nodes may also be present and can be the first manifestation of the disease.[17][21] Rashes occur early in the disease, and the cutaneous rash observed in patients with KD is non-specific, polymorphic, non-itchy and normally observed up to the 5th day of fever. Cutaneous exanthema may comprise macular-papular erythematous and fissure lesions, the most common type, in addition to urticariform type rash, purpuric, multiform-like erythema.[22] and peeling of the skin in the genital area, hands, and feet (especially around the nails and on the palms and soles) may occur in later phases.[17][23] Some of these symptoms may come and go during the course of the illness. It is a syndrome affecting multiple organ systems, and in the acute stage of KD, systemic inflammatory changes are evident in many organs.[9] Myocarditis,[24] pericarditis, valvulitis, aseptic meningitis, pneumonitis, lymphadenitis, and hepatitis may be present and are manifested by the presence of inflammatory cells in the affected tissues.[9] If left untreated, some symptoms will eventually relent, but coronary artery aneurysms will not improve, resulting in a significant risk of death or disability due to myocardial infarction (heart attack).[12] If treated in a timely fashion, this risk can be mostly avoided and the course of illness cut short.[25]
| Less common manifestations | |
|---|---|
| System | Manifestations |
| GIT | Diarrhea, abdominal pain, vomiting, liver dysfunction, pancreatitis, Hydrops gallbladder, cholangitis, intussusception, intestinal pseudo-obstruction, ascites, splenic infarction. |
| MSS | Polyarthritis and arthralgia. |
| CVS | Myocarditis, pericarditis, valvular heart disease. |
| GU | Urethritis, prostatitis, cystitis, priapism, Interstitial nephritis, orchitis, nephrotic syndrome. |
| CNS | Aseptic meningitis, and sensorineural deafness. |
| RS | Influenza-like illness, plural effusion, Atelectasis. |
| Skin | Erythema and induration at BCG vaccine site, Beau's lines, and finger gangrene. |
| Source: review,[12] table.[26] | |
- High-grade fever (greater than 39 °C or 102 °F; often as high as 40 °C or 104 °F),[12] The duration of fever is on average one to two weeks; in the absence of treatment, it may extend for three to four weeks.[12] However, when appropriate therapy is started the fever is gone after two days.[17]
- Red eyes (conjunctivitis) bilateral without pus or drainage, also known as "conjunctival injection".[19]
- Anterior uveitis.[19]
- Bright red, chapped, or cracked lips.[12]
- Red mucous membranes in the mouth.[12]
- Strawberry tongue, white coating on the tongue or prominent red bumps (papillae) on the back of the tongue.[12]
- Red palms of the hands and the soles of the feet.[12]
- Peeling (desquamation) palms and soles (later in the illness); peeling may begin around the nails.[4][17]
- Rash which may take many forms, non-specific, polymorphic, non-itchy, but not vesicle-bullous lesions, and appears on the trunk.[12]
- Swollen lymph nodes (frequently only one lymph node is swollen, and is usually on onc side), particularly in the neck area.[23]
- Joint pain (arthralgia) and swelling, frequently symmetrical, Also arthritis can occur.[12]
- Irritability.[12]
- Tachycardia (rapid heart beat).[12]
- Beau's lines (transverse grooves on nails).[12]
- May find breathing difficult.[12]
Complications
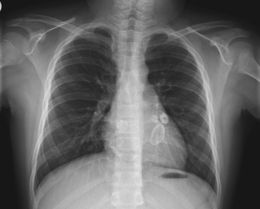
The cardiac complications are the most important aspect of the disease. Kawasaki disease can cause vasculitic changes (inflammation of blood vessels) in the coronary arteries and subsequent coronary artery aneurysms. These aneurysms can lead to myocardial infarction (heart attack) even in young children. Overall, about 10–18% of children with Kawasaki disease develop coronary artery aneurysms with much higher prevalence among patients who are not treated early in the course of illness. Kawasaki disease and rheumatic fever are the most common causes of acquired heart disease among children in the United States.[27][28]
Causes
Like all autoimmune diseases, the cause of Kawasaki disease is presumably the interaction of genetic and environmental factors, possibly including an infection. The specific cause is unknown,[29][30][31] but current theories center primarily on immunological causes for the disease. Evidence increasingly points to an infectious etiology,[32] but debate continues on whether the cause is a conventional antigenic substance or a superantigen.[33] Children's Hospital Boston reports that "[s]ome studies have found associations between the occurrence of Kawasaki disease and recent exposure to carpet cleaning or residence near a body of stagnant water; however, cause and effect have not been established."[28]
An association has been identified with a SNP in the ITPKC gene, which codes an enzyme that negatively regulates T-cell activation.[34] An additional factor that suggests genetic susceptibility is the fact that regardless of where they are living, Japanese children are more likely than other children to contract the disease.[28] The HLA-B51 serotype has been found to be associated with endemic instances of the disease.[35]
Diagnosis
| Criteria for Diagnosis of Kawasaki Disease |
|---|
| Fever of ≥5 days' duration associated with at least 4† of the following 5 changes |
| Bilateral nonsuppurative conjunctivitis |
| One of more changes of the mucous membranes of the upper respiratory tract, including pharyngeal injection, dry fissured lips, injected lips, and "strawberry" tongue |
| One or more changes of the extremities, including peripheral erythema, peripheral edema, periungual desquamation, and generalized desquamation |
| Polymorphous rash, primarily truncal |
| Cervical lymphadenopathy >1.5 cm in diameter |
| Disease cannot be explained by some other known disease process |
| †A diagnosis of Kawasaki disease can be made if fever and only 3 changes are present in conjunction with coronary artery disease documented by two-dimensional echocardiography or coronary angiography. |
| Source: Nelson's essentials of pediatrics[36], Review[37] |
Kawasaki disease can only be diagnosed clinically (i.e. by medical signs and symptoms). There exists no specific laboratory test for this condition. It is difficult to establish the diagnosis, especially early in the course of the illness, and frequently children are not diagnosed until they have seen several health care providers. Many other serious illnesses can cause similar symptoms, and must be considered in the differential diagnosis, including scarlet fever, toxic shock syndrome, juvenile idiopathic arthritis, and childhood mercury poisoning (acrodynia).
Classically, five days of fever[38] plus four of five diagnostic criteria must be met in order to establish the diagnosis. The criteria are: (1) erythema of the lips or oral cavity or cracking of the lips; (2) rash on the trunk; (3) swelling or erythema of the hands or feet; (4) red eyes (conjunctival injection) (5) swollen lymph node in the neck of at least 15 millimeters.
Many children, especially infants, eventually diagnosed with Kawasaki disease do not exhibit all of the above criteria. In fact, many experts now recommend treating for Kawasaki disease even if only three days of fever have passed and at least three diagnostic criteria are present, especially if other tests reveal abnormalities consistent with Kawasaki disease. In addition, the diagnosis can be made purely by the detection of coronary artery aneurysms in the proper clinical setting.
Investigations
A physical examination will demonstrate many of the features listed above.
Blood tests
- Complete blood count (CBC) may reveal normocytic anemia and eventually thrombocytosis
- Erythrocyte sedimentation rate (ESR) will be elevated
- C-reactive protein (CRP) will be elevated
- Liver function tests may show evidence of hepatic inflammation and low serum albumin
Other optional tests
- Electrocardiogram may show evidence of ventricular dysfunction or, occasionally, arrhythmia due to myocarditis
- Echocardiogram may show subtle coronary artery changes or, later, true aneurysms.
- Ultrasound or computerized tomography may show hydrops (enlargement) of the gallbladder
- Urinalysis may show white blood cells and protein in the urine (pyuria and proteinuria) without evidence of bacterial growth
- Lumbar puncture may show evidence of aseptic meningitis
- Angiography was historically used to detect coronary artery aneurysms and remains the gold standard for their detection, but is rarely used today unless coronary artery aneurysms have already been detected by echocardiography.
Treatment
Children with Kawasaki disease should be hospitalized and cared for by a physician who has experience with this disease. When in an academic medical center, care is often shared between pediatric cardiology and pediatric infectious disease specialists (although no specific infectious agent has been identified yet).[28] It is imperative that treatment be started as soon as the diagnosis is made to prevent damage to the coronary arteries.
Intravenous immunoglobulin (IVIG) is the standard treatment for Kawasaki disease[39] and is administered in high doses with marked improvement usually noted within 24 hours. If the fever does not respond, an additional dose may have to be considered. In rare cases, a third dose may be given to the child. IVIG by itself is most useful within the first seven days of onset of fever, in terms of preventing coronary artery aneurysm.
Salicylate therapy, particularly aspirin, remains an important part of the treatment (though questioned by some)[40] but salicylates alone are not as effective as IVIG. Aspirin therapy is started at high doses until the fever subsides, and then is continued at a low dose when the patient returns home, usually for two months to prevent blood clots from forming. Except for Kawasaki disease and a few other indications, aspirin is otherwise normally not recommended for children due to its association with Reye's syndrome. Because children with Kawasaki disease will be taking aspirin for up to several months, vaccination against varicella and influenza is required, as these infections are most likely to cause Reye's syndrome.[41]
Corticosteroids have also been used,[42] especially when other treatments fail or symptoms recur, but in a randomized controlled trial, the addition of corticosteroid to immune globulin and aspirin did not improve outcome.[43] In cases of kawasaki disease refractory to IVIG, cyclophosphamide and plasma exchange have been investigated as possible treatments, with variable outcomes.
There are also treatments for iritis and other eye symptoms. Another treatment may include the use of Infliximab (Remicade). Infliximab works by binding tumour necrosis factor alpha.
Prognosis
With early treatment, rapid recovery from the acute symptoms can be expected and the risk of coronary artery aneurysms greatly reduced. Untreated, the acute symptoms of Kawasaki disease are self-limited (i.e. the patient will recover eventually), but the risk of coronary artery involvement is much greater. Overall, about 2% of patients die from complications of coronary vasculitis. Patients who have had Kawasaki disease should have an echocardiogram initially every few weeks, and then every one or two years to screen for progression of cardiac involvement.
It is also not uncommon that a relapse of symptoms may occur soon after initial treatment with IVIG. This usually requires re-hospitalization and re-treatment. Treatment with IVIG can cause allergic and non-allergic acute reactions, aseptic meningitis, fluid overload and, rarely, other serious reactions. Overall, life-threatening complications resulting from therapy for Kawasaki disease are exceedingly rare, especially compared with the risk of non-treatment.
Epidemiology
By far the highest incidence of Kawasaki disease occurs in Japan (175 per 100,000), though its incidence in the United States is increasing. Kawasaki disease is predominantly a disease of young children, with 80% of patients younger than five years of age. The disease affects boys more than girls. Kawasaki was extremely uncommon in caucasians until the last few decades. Approximately 2,000-4,000 cases are identified in the United States each year.[27][28]
See also
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 1232–4. ISBN 1-4160-2999-0.
- ↑ "Merck Manual, Online edition: Kawasaki Disease". http://www.merck.com/mmpe/sec19/ch286/ch286d.html. Retrieved May 9, 2010.
- ↑ Okano M, Luka J, Thiele GM, Sakiyama Y, Matsumoto S, Purtilo DT (October 1989). "Human herpesvirus 6 infection and Kawasaki disease". Journal of Clinical Microbiology 27 (10): 2379–80. PMID 2555393. PMC 267029. http://jcm.asm.org/cgi/pmidlookup?view=long&pmid=2555393.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 Kim DS (December 2006). "Kawasaki disease". Yonsei Medical Journal 47 (6): 759–72. PMID 17191303. PMC 2687814. http://www.eymj.org/DOIx.php?id=10.3349/ymj.2006.47.6.759.
- ↑ Kawasaki T (1967). "[Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]" (in Japanese). Arerugi 16 (3): 178–222. PMID 6062087.
- ↑ 6.0 6.1 6.2 6.3 6.4 Guillevin L, Pagnoux C (Mar 2008). "[Classification of systemic vasculitides]" (in French). La Revue Du Praticien 58 (5): 480–6. PMID 18524103.
- ↑ http://medical-dictionary.thefreedictionary.com/necrotizing+vasculitis
- ↑ Dillon MJ, Eleftheriou D, Brogan PA (Nov 2009). "Medium-size-vessel vasculitis". Pediatric Nephrology (Berlin, Germany). doi:10.1007/s00467-009-1336-1. PMID 19946711. http://dx.doi.org/10.1007/s00467-009-1336-1.
- ↑ 9.0 9.1 9.2 Fujiwara H, Fujiwara T, Kao TC, Ohshio G, Hamashima Y (Jun 1986). "Pathology of Kawasaki disease in the healed stage. Relationships between typical and atypical cases of Kawasaki disease". Acta Pathologica Japonica 36 (6): 857–67. PMID 3766134.
- ↑ Rigante D (2006). "Clinical overview of vasculitic syndromes in the pediatric age". European Review for Medical and Pharmacological Sciences 10 (6): 337–45. PMID 17274537.
- ↑ Brandt HR, Arnone M, Valente NY, Sotto MN, Criado PR (2009). "[Medium and large vessel vasculitis"] (in Portuguese). An Bras Dermatol 84 (1): 55–67. PMID 19377760. http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0365-05962009000100008&lng=en&nrm=iso&tlng=en.
- ↑ 12.00 12.01 12.02 12.03 12.04 12.05 12.06 12.07 12.08 12.09 12.10 12.11 12.12 12.13 12.14 12.15 Castro PA, Urbano LM, Costa IM (Aug 2009). "[Kawasaki disease"] (in Portuguese). Anais Brasileiros De Dermatologia 84 (4): 317–29. PMID 19851663. http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0365-05962009000400002&lng=en&nrm=iso&tlng=en.
- ↑ Herlin T, Nielsen S (Sep 2008). "[Primary childhood vasculitis--new classification criteria]". Ugeskr Laeger 170 (36): 2784–2787. PMID 18761873.
- ↑ 14.0 14.1 14.2 PMID 16322081 (PubMed)
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand - ↑ Guillevin L, Pagnoux C, Guilpain P (May 2007). "[Classification of systemic vasculatides"] (in French). Presse Med 36 (5 Pt 2): 845–53. doi:10.1016/j.lpm.2007.01.035. PMID 17408915. http://www.masson.fr/masson/S0755-4982(07)00189-3.
- ↑ Jennette JC, Falk RJ (Oct 2000). "Do vasculitis categorization systems really matter?". Curr Rheumatol Rep 2 (5): 430–8. PMID 11123094.
- ↑ 17.0 17.1 17.2 17.3 17.4 17.5 Rowley AH, Shulman ST (Jul 1998). "Kawasaki syndrome". Clinical Microbiology Reviews 11 (3): 405–14. PMID 9665974. PMC 88887. http://cmr.asm.org/cgi/pmidlookup?view=long&pmid=9665974.
- ↑ 18.0 18.1 Kawasaki T (Jan 1995). "General review and problems in Kawasaki disease". Japanese Heart Journal 36 (1): 1–12. PMID 7760506.
- ↑ 19.0 19.1 19.2 Smith LB, Newburger JW, Burns JC (Feb 1989). "Kawasaki syndrome and the eye". The Pediatric Infectious Disease Journal 8 (2): 116–8. PMID 2468129.
- ↑ Bachmeyer C, Turc Y, Curan D, Duval-Arnould M (Jan 2000). "Anterior uveitis as the initial sign of adult Kawasaki syndrome (mucocutaneous lymph node syndrome)". American Journal of Ophthalmology 129 (1): 101–2. PMID 10653425. http://linkinghub.elsevier.com/retrieve/pii/S0002939499002858.
- ↑ Kubota M, Usami I, Yamakawa M, Tomita Y, Haruta T (Jun 2008). "Kawasaki disease with lymphadenopathy and fever as sole initial manifestations". Journal of Paediatrics and Child Health 44 (6): 359–62. doi:10.1111/j.1440-1754.2008.01310.x. PMID 18476929. http://www3.interscience.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=1034-4810&date=2008&volume=44&issue=6&spage=359.
- ↑ Dajani AS, Taubert KA, Gerber MA, et al. (May 1993). "Diagnosis and therapy of Kawasaki disease in children". Circulation 87 (5): 1776–80. PMID 8491037. http://circ.ahajournals.org/cgi/pmidlookup?view=long&pmid=8491037.
- ↑ 23.0 23.1 Chung CJ, Stein L (Jul 1998). "Kawasaki disease: a review". Radiology 208 (1): 25–33. PMID 9646789. http://radiology.rsnajnls.org/cgi/pmidlookup?view=long&pmid=9646789.
- ↑ Dahdah N (Apr 2010). "Not just coronary arteritis, Kawasaki disease is a myocarditis, too". Journal of the American College of Cardiology 55 (14): 1507; author reply 1507–8. doi:10.1016/j.jacc.2009.11.067. PMID 20359606. http://linkinghub.elsevier.com/retrieve/pii/S0735-1097(10)00398-0.
- ↑ Tse SM, Silverman ED, McCrindle BW, Yeung RS (Apr 2002). "Early treatment with intravenous immunoglobulin in patients with Kawasaki disease". J. Pediatr. 140 (4): 450–5. doi:10.1067/mpd.2002.122469. PMID 12006960. http://linkinghub.elsevier.com/retrieve/pii/S0022-3476(02)76010-1.
- ↑ http://www.scielo.br/img/revistas/abd/v84n4/en_4a02qua03.jpg
- ↑ 27.0 27.1 "Kawasaki Disease - Signs and Symptoms". http://www.ucsfchildrenshospital.org/conditions/kawasaki_disease/signs_and_symptoms.html.
- ↑ 28.0 28.1 28.2 28.3 28.4 "Who Kawasaki Disease Affects". Children's Hospital Boston. http://www.childrenshospital.org/clinicalservices/Site468/mainpageS468P5.html. Retrieved 2009-01-04.
- ↑ Rowley AH, Baker SC, Orenstein JM, Shulman ST (May 2008). "Searching for the cause of Kawasaki disease--cytoplasmic inclusion bodies provide new insight". Nat. Rev. Microbiol. 6 (5): 394–401. doi:10.1038/nrmicro1853. PMID 18364728.
- ↑ "Kawasaki Disease". American Heart Association. http://www.americanheart.org/presenter.jhtml?identifier=4634. Retrieved 3 January 2009.
- ↑ "Kawasaki Disease: Causes". Mayo Clinic. http://www.mayoclinic.com/health/kawasaki-disease/DS00576/DSECTION=causes. Retrieved 3 January 2009.
- ↑ Nakamura Y, Yashiro M, Uehara R, Oki I, Watanabe M, Yanagawa H (2008). "Monthly observation of the number of patients with Kawasaki disease and its incidence rates in Japan: chronological and geographical observation from nationwide surveys" (– Scholar search). J Epidemiol 18 (6): 273–9. doi:10.2188/jea.JE2008030. PMID 19075496. http://joi.jlc.jst.go.jp/JST.JSTAGE/jea/JE2008030?from=PubMed.
- ↑ Freeman AF, Shulman ST (June 2001). "Recent developments in Kawasaki disease". Curr Opin Infect Dis 14 (3): 357–61. PMID 11964855.
- ↑ Onouchi Y, Gunji T, Burns JC, et al. (January 2008). "ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms". Nat. Genet. 40 (1): 35–42. doi:10.1038/ng.2007.59. PMID 18084290.
- ↑ Keren G, Danon YL, Orgad S, Kalt R, Gazit E (August 1982). "HLA Bw51 is increased in mucocutaneous lymph node syndrome in Israeli patients". Tissue Antigens 20 (2): 144–6. PMID 6958087.
- ↑ Behrman, Richard E.; Kliegman, Robert; Karen Marcdante; Jenson, Hal B. (2006). Nelson essentials of pediatrics. St. Louis, Mo: Elsevier Saunders. ISBN 1-4160-0159-X.
- ↑ Ozen S, Ruperto N, Dillon MJ, et al. (July 2006). "EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides". Ann. Rheum. Dis. 65 (7): 936–41. doi:10.1136/ard.2005.046300. PMID 16322081. PMC 1798210. http://ard.bmj.com/cgi/pmidlookup?view=long&pmid=16322081.
- ↑ "Kawasaki Disease - June 1999 - American Academy of Family Physicians". http://www.aafp.org/afp/990600ap/3093.html.
- ↑ Oates-Whitehead RM, Baumer JH, Haines L, et al. (2003). "Intravenous immunoglobulin for the treatment of Kawasaki disease in children". Cochrane Database Syst Rev (4): CD004000. doi:10.1002/14651858.CD004000. PMID 14584002.
- ↑ Hsieh KS, Weng KP, Lin CC, Huang TC, Lee CL, Huang SM (December 2004). "Treatment of acute Kawasaki disease: aspirin's role in the febrile stage revisited". Pediatrics 114 (6): e689–93. doi:10.1542/peds.2004-1037. PMID 15545617. http://pediatrics.aappublications.org/cgi/pmidlookup?view=long&pmid=15545617.
- ↑ http://emedicine.medscape.com/article/804960-treatment
- ↑ Sundel RP, Baker AL, Fulton DR, Newburger JW (June 2003). "Corticosteroids in the initial treatment of Kawasaki disease: report of a randomized trial". J. Pediatr. 142 (6): 611–6. doi:10.1067/mpd.2003.191. PMID 12838187. http://linkinghub.elsevier.com/retrieve/pii/S0022347603001173.
- ↑ Newburger JW et al., Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease, N Engl J Med. 2007 Feb 25;356(7):663-75
{kind=link}
External links
- Kawasaki Disease Foundation
- Kawasaki Disease Canada
- Kawasaki Disease Forum
- Kawasaki Disease Research Program
- Kawasaki Disease information from Seattle Children's Hospital Heart Center
- Kawasaki Disease, MedlinePlus
- Australian Kawasaki Support Network
- Kawasaki Disease Foundation of South Africa
|
||||||||||||||||||||||||||||